The glycogen storage disease type II (GSD II) is a rare, multisystemic disease with variable rates of disease progression that is also called Pompe disease. It is caused by alterations in the acid α-glucosidase (GAA) gene, resulting in reduced levels of this enzyme that in turn leads to a reduced degradation and thus accumulation of glycogen mostly in different muscle tissues. Most common symptoms include cardiac dysfunction, muscle weakness, hypotonia, respiratory problems and a strongly reduced life expectancy.
The 6neo mouse model has a Gaa knockout resulting in reduced GAA enzyme levels and thus a progressive accumulation of glycogen in different muscle tissues.
Based on published data, mice present the following pathologies starting at 3 weeks of age:
- Reduced GAA activity in brain, spinal cord, liver, and muscle
- Glycogen accumulations in brain, spinal cord, heart, skeletal muscle, and diaphragm (PAS reaction)
Behavioral analyses performed in-house verified strong muscle weakness as well as motor deficits as analyzed by the grip strength test and beam walk test.
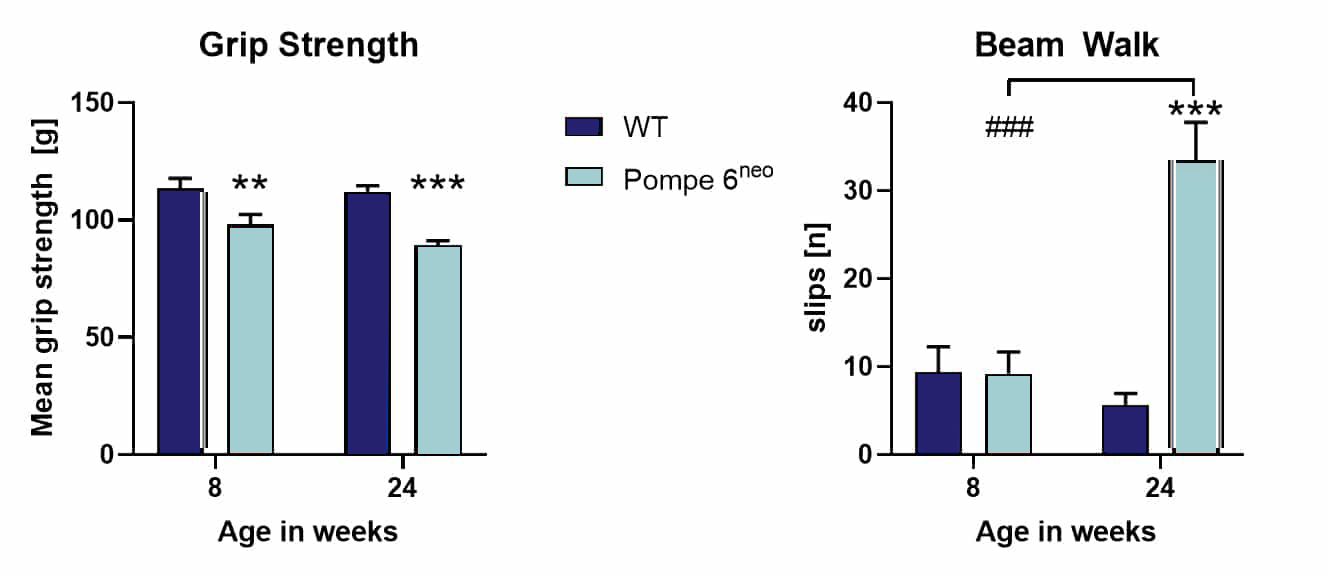
Figure: Grip strength test and beam walk test of 6neo mice. Mean grip strength (A) and number of slips (B) of 6neo mice and WT controls at the age of 8 and 24 weeks; 8 weeks: n = 24 per group; 24 weeks: n = 48 per group. Two-way ANOVA with Bonferroni’s post hoc test; mean + SEM; ***/###p<0.001; ***differences between genotypes; ###differences between age groups.
Scantox offers a custom-tailored study design for this model, and we are flexible to accommodate to your special interest. 6neo mice show relevant features of Pompe disease already at young age. This allows for extraordinarily fast turn-around times.
We would be happy to test your compounds in the 6neo mouse model! The most common readouts are:
- Enzyme levels in neuronal and visceral tissues
- Substrate levels in neuronal and visceral tissues
- Muscle weakness and motor deficits
You might be also interested in these related topics:
- 4L/PS-NA Gaucher disease mouse model
- CBE-induced Gaucher disease mouse model
- GBA D409V KI mouse model